شركة رقمي
نقل من موقع 

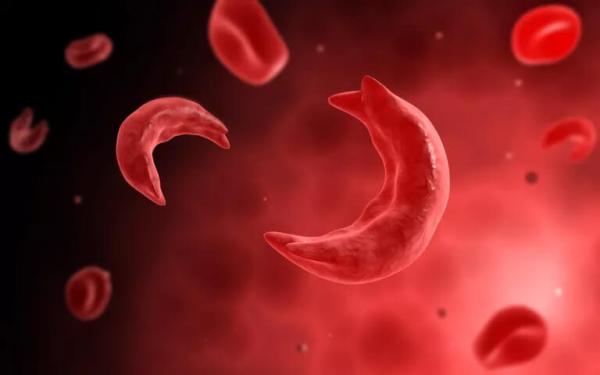
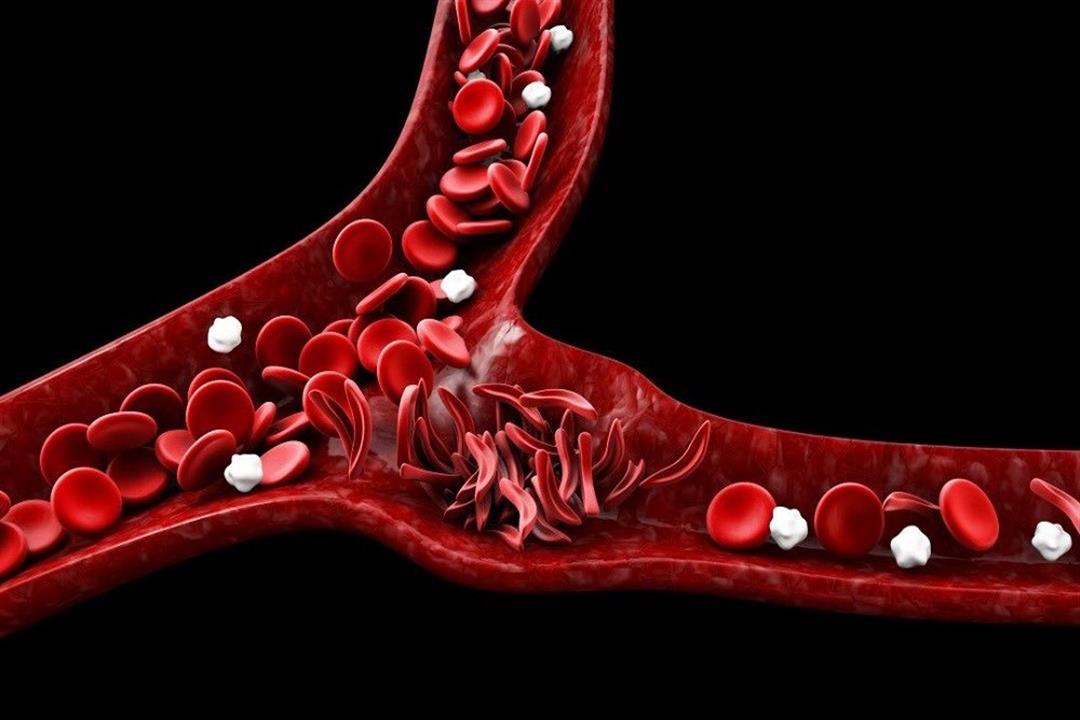
قال الدكتور ربيع حنّا، أخصائي أمراض الدم وأورام الأطفال في مستشفى كليفلاند كلينك للأطفال، إن التعامل مع مرض فقر الدم المنجلي يشهد حاليا تطورات لافتة ومثيرة، لاسّيما مع التقدم الذي تم تحقيقه في مجال الأبحاث وتطوير علاجات جديدة من شأنها منح الملايين من المرضى حول العالم أملا حقيقيا بالتعافي. وأضاف حنّا أن مرض فقر الدم المنجلي يمكن أن يؤثر بشدة على جودة حياة المرضى ويحد من إمكاناتهم بسبب نوبات الألم الشديدة وتلف الأعضاء الطرفية وانخفاض متوسط العمر المتوقع؛ حيث يمكن للأدوية تخفيف شدة المرض وعلاج الأعراض التي يسببها، ولكن متوسط عمر المريض يكون عادة في منتصف الأربعينات، ولهذا من المهم للغاية الوصول إلى علاج فعّال لهذا المرض. 300 ألف طفل يولدون سنويا مع اضطرابات شديدة في الهيموغلوبين ويتسبب التغير الجيني في الحمض النووي لدى الأشخاص المصابين بهذا المرض في حدوث تغير كيميائي في الهيموغلوبين، وهو بروتين أحمر مسؤول عن نقل الأكسجين في الدم، ما يؤدي إلى تحوّل الخلايا إلى ما يشبه شكل “المنجل” بدلا من شكلها الطبيعي المستدير، الأمر الذي يعيق مرورها بسهولة عبر الأوعية الدموية، كما يمكنها أيضا أن تسدَّ الأوعية أو تتفكك، ما يؤدي إلى انخفاض عمر خلايا الدم الحمراء وزيادة معدل تخزين الحديد في الكبد والقلب. ويمكن أن يؤدي ذلك إلى إصابة المريض بمضاعفات خطيرة مثل تليف الكبد وفشل الكبد والسكتة الدماغية واعتلال عضلة القلب وفشل القلب، هذا بالإضافة إلى الآلام الشديدة. وتشمل الخيارات العلاجية للمصابين بهذا المرض عمليات زراعة الدم أو النخاع، ولكن في الواقع من الصعب العثور على متبرع متطابق كما أن هناك مخاطر كبيرة مرتبطة بهذا النوع من العلاجات. وأوضح الدكتور حنّا هذه المخاطر قائلا “أولا، تتراوح نسبة مخاطر عدم تقبل جسم المريض للخلايا الممنوحة بين 5 في المئة و10 في المئة. ثانيا، هناك خطر الإصابة بمرض ‘الطعم حيال المضيف’ والذي يحدث عندما يهاجم نخاع عظم المتبرع أو الخلايا الجذعية للمتلقي. ولتفادي مثل هذه الحالة، يتم إعطاء المريض أدوية قوية مثبطة للمناعة، ولكن لا يزال هناك خطر كبير للإصابة بمرض الطعم حيال المضيف”. وأضاف “في المقابل يعتمد العلاج الجيني على الخلايا الجذعية الخاصة بالمريض، وبالتالي، يتم التخلص من مخاطر عدم تقبل المريض للخلايا أو احتمالية الإصابة بمرض الطعم حيال المضيف”. ربيع حنّا: مرض فقر الدم المنجلي يمكن أن يؤثر بشدة على جودة حياة المرضى ومن الفوائد الأخرى للعلاج الجيني أن المرضى لن يحتاجوا إلى أدوية مثبطة للمناعة؛ لذا سيعمل جهاز المناعة لديهم بشكل طبيعي، وهو أمر مهم بشكل خاص خلال الجائحة. بالإضافة إلى ذلك، وبينما يحتاج المرضى إلى العلاج الكيميائي لتعزيز جهوزية الجسم سواء عند إجراء عمليات زراعة تقليدية أو عمليات العلاج الجيني، فإن العلاج الكيميائي المستخدم في العلاج الجيني يكون أقل قوة. وتابع الدكتور حنّا “يمثل العلاج الجيني تقنية رائدة تعمل عن طريق استبدال أو تعطيل الجينات المسببة للأمراض. ونقوم في تجربتنا السريرية التي نجريها حاليا بإدخال جينات صحية وسليمة في الجسم بهدف تصحيح التشوهات الجينية لخلايا الدم الحمراء. ومن خلال تمكين الخلايا من إنتاج المزيد من الهيموغلوبين الجيني، فإن هذا العلاج لديه القدرة على علاج مرض فقر الدم المنجلي بطريقة دقيقة”. ومن الأدوية التي تمت الموافقة عليها من جانب إدارة الغذاء والدواء الأميركية خلال عامي 2019 و2021 على التوالي، دواء “كريزانليزيماب”، والذي يعمل على تقليل الآلام والمضاعفات الشديدة، ودواء “فوكسيلوتور”، والذي يساعد على استعادة الوظائف الطبيعية لخلايا الدم الحمراء وتوصيل الأكسجين إلى الأنسجة، مما يساعد بالتالي على تخفيف الألم. ووفقا لبيانات منظمة الصحة العالمية، فإن نحو 5 في المئة من سكان العالم يحملون جينات السمات المسببة لاضطرابات الهيموغلوبين، وخاصة مرض فقر الدم المنجلي والثلاسيميا. ومع ذلك تجدر الإشارة إلى أن هذا لا يعني أنهم سيصابون بالمرض. وتشير التقديرات إلى أن 300 ألف طفل يولدون سنويا مع اضطرابات شديدة في الهيموغلوبين. وتعتبر الهند وبعض بلدان الشرق الأوسط وأفريقيا من بين تلك التي ينتشر فيها مرض فقر الدم المنجلي.
مشاركة :